refdb_folder <- here::here("data", "refdb")
refdb_folder[1] "/home/rstudio/mydatalocal/course-material/data/refdb"To begin with, download the course repository on your computer or virtual machine.
To do so, visit the (ANF-metabarcoding) repository on github and download it as a zip file.
You can also download the repository directly from the terminal using this commmand:
wget https://github.com/ANF-MetaBioDiv/course-material/archive/refs/heads/main.zipOnce on your machine, unzip the file and go to the resulting folder or place the resulting folder in the most convenient location (setwd("~/course-material-main") for example).
💡 Tip: Work within an RStudio project. Open your course-material-main folder as an RStudio project (create or use a .Rproj file inside it). Then here::here(“data”, “refdb”) will correctly point to /home/rstudio/Documents/course-material-main/data/refdb.
Save as a variable the path to the folder where you will place the references databases.
refdb_folder <- here::here("data", "refdb")
refdb_folder[1] "/home/rstudio/mydatalocal/course-material/data/refdb"In this course we use as much as possible the :: operator. It allows calling a function from a given package without having to load the entire package using library(package). This way it is clear where the functions come from. In that case, the function here() comes from the package here.
The reason why we use here::here() is that when you render a Rmarkdown file, the working directory is where the Rmarkdown file is:
getwd()Whereas here::here() point to the root of the R project
here::here()Now, let’s create the folder directly from R:
if (!dir.exists(refdb_folder)) dir.create(refdb_folder, recursive = TRUE)You can also create the folder from RStudio in the Files window
💡 Tip: You can access the documentation of any R function using ? in the console. If you to know everything about the function dir.create(), simply run ?dir.create()
The Silva reference database, commonly used to assign 16S metabarcoding data, will be used in practical.
In case you are working with 18S sequences, you will have better assignments using PR2 or EukRibo especially if you are interested in protists.
The following code downloads dada2 formatted silva reference databases. If you are not comfortable with it, you can simply download the reference database files from your web browser here.
# R stop downloading after timeout which is
# 60 seconds by default
getOption("timeout")[1] 60# so we change timeout to be 20 minutes
options(timeout = 1200)
# we save in variable the path to the refdb
# in the working space
silva_train_set <-
file.path(
refdb_folder,
"silva_nr99_v138.2_toGenus_trainset.fa.gz"
)
silva_species_assignment <-
file.path(
refdb_folder,
"silva_v138.2_assignSpecies.fa.gz"
)
# then we download the files if they don't already exist
if (!file.exists(silva_train_set)) {
download.file(
"https://zenodo.org/records/14169026/files/silva_nr99_v138.2_toGenus_trainset.fa.gz",
silva_train_set,
quiet = TRUE
)
}
if (!file.exists(silva_species_assignment)) {
download.file(
"https://zenodo.org/records/14169026/files/silva_v138.2_assignSpecies.fa.gz",
silva_species_assignment,
quiet = TRUE
)
}We will use in this practical R functions especially written for this course. The “classic” way to import functions is to use source() with the name of the R script to source.
Instead, we use devtools::load_all(). This function will source all the scripts from the folder R/ along with the documentation in man/ :
devtools::load_all()Warning: Objects listed as exports, but not present in namespace:
• primer_trimSave the path to the directory containing your raw data (paired-end sequencing fastq files) in a variable named path_to_fastqs.
path_to_fastqs <- here::here("data", "raw")The gzipped (compressed) FASTQ formatted “forward” (R1) and “reverse” (R2) files are named as follow:
${SAMPLENAME}_R1.fastq.gz for the forward files${SAMPLENAME}_R2.fastq.gz for the reverse files.We list the forward files using the function list.files(). The argument pattern gives you the possibility to only select file names matching a regular expression. In our case, we select all file names finishing by _R1.fastq.gz.
fnFs <- sort(list.files(path_to_fastqs,
pattern = "_R1.fastq.gz",
full.names = TRUE))We do the same for reverse samples.
fnRs <- sort(list.files(path_to_fastqs,
pattern = "_R2.fastq.gz",
full.names = TRUE))To understand: What fnFs & fnRs variables contain?
sample_names <- basename(fnFs) |>
strsplit(split = "_") |>
sapply(head, 1)To understand:
basename(): remove path to only keep file name.
|>: R “pipe”. It allows you to chain functions, avoiding intermediate variables and nested parenthesis. It basically transfers the output of the left expression to the input of the right expression. You need R > 4.1 to use this pipe, otherwise use %>% from magrittr.
strsplit(): split character chain according to a defined pattern. ?strsplit for documentation.
sapply(): apply a function to each element of a list or vector. The output is simplified to be vector.
Let’s go step by step. First list the R1 file names.
basename(fnFs) |>
head()[1] "S11B_R1.fastq.gz" "S1B_R1.fastq.gz" "S2B_R1.fastq.gz" "S2S_R1.fastq.gz"
[5] "S3B_R1.fastq.gz" "S3S_R1.fastq.gz" We can see that the sample name is before the first _. With strsplit(), we can split each file name into a 2 elements vector. The result is a list of 2 elements vectors.
basename(fnFs) |>
strsplit(split = "_") |>
head()[[1]]
[1] "S11B" "R1.fastq.gz"
[[2]]
[1] "S1B" "R1.fastq.gz"
[[3]]
[1] "S2B" "R1.fastq.gz"
[[4]]
[1] "S2S" "R1.fastq.gz"
[[5]]
[1] "S3B" "R1.fastq.gz"
[[6]]
[1] "S3S" "R1.fastq.gz"Now, We just have to extract the first element for each file.
basename(fnFs) |>
strsplit(split = "_") |>
sapply(head, 1) |>
head()[1] "S11B" "S1B" "S2B" "S2S" "S3B" "S3S" Tip: you can achieve the same thing using regular expressions:
gsub("^.+/|_.+$", "", fnFs) |> head()[1] "S11B" "S1B" "S2B" "S2S" "S3B" "S3S" Regular expressions are extremely useful. If you are keen to learn how to use them, have a look here
We use a custom function, qualityprofile(), implemented in R/preprocessing.R to check the quality of the raw sequences.
Run ?qualityprofile to know more about this function.
# create a directory for the outputs
quality_folder <- here::here("outputs",
"dada2",
"quality_plots")
if (!dir.exists(quality_folder)) {
dir.create(quality_folder, recursive = TRUE)
}
qualityprofile(fnFs,
fnRs,
file.path(quality_folder, "quality_plots.pdf"))Open the the pdf file generated by qualityprofile()
We first create a folder where to save the reads once they are trimmed:
path_to_trimmed_reads <- here::here(
"outputs",
"dada2",
"trimmed"
)
if (!dir.exists(path_to_trimmed_reads)) dir.create(path_to_trimmed_reads, recursive = TRUE)Then we prepare a list of paths where to save the results (e.g. sequences without primers)
nopFw <- file.path(path_to_trimmed_reads, basename(fnFs))
nopRv <- file.path(path_to_trimmed_reads, basename(fnRs))
head(nopFw)[1] "/home/rstudio/mydatalocal/course-material/outputs/dada2/trimmed/S11B_R1.fastq.gz"
[2] "/home/rstudio/mydatalocal/course-material/outputs/dada2/trimmed/S1B_R1.fastq.gz"
[3] "/home/rstudio/mydatalocal/course-material/outputs/dada2/trimmed/S2B_R1.fastq.gz"
[4] "/home/rstudio/mydatalocal/course-material/outputs/dada2/trimmed/S2S_R1.fastq.gz"
[5] "/home/rstudio/mydatalocal/course-material/outputs/dada2/trimmed/S3B_R1.fastq.gz"
[6] "/home/rstudio/mydatalocal/course-material/outputs/dada2/trimmed/S3S_R1.fastq.gz" The data you are working with correspond to the V3-V4 region using the primers Pro341F (CCTACGGGNBGCASCAG) and Pro805R (GACTACNVGGGTATCTAAT). Save into variables the forward and reverse primers:
primer_fwd <- "CCTACGGGNBGCASCAG"
primer_rev <- "GACTACNVGGGTATCTAAT"Let’s have a closer look at sequences and find the primers.
Forward reads would contain CCTACGGGNBGCASCAG
Biostrings::readDNAStringSet(
fnFs[1],
format = "fastq",
nrec = 10
)DNAStringSet object of length 10:
width seq names
[1] 293 CCTACGGGGGGCAGCAGTAGGGA...ACATCGGCTTAACCGATGAAGT M01522:260:000000...
[2] 293 CCTACGGGTGGCACCAGTAGGGA...CGGGGCTTAACCTCGGAACTGC M01522:260:000000...
[3] 292 CCTACGGGGCGCAGCAGGCGCGA...GGGACCGGGAGAGGTGTGAGGT M01522:260:000000...
[4] 293 CCTACGGGGTGCAGCAGTAGGGA...TCAAAACTCCCAGTCTAGAGTT M01522:260:000000...
[5] 291 CCTACGGGTGGCAGCAGTGGGGA...GCAGTGGAAACTGTTGGGCTTG M01522:260:000000...
[6] 293 CCTACGGGATGCAGCAGGCGCGA...GGGACCGGGAGAGGTGTGGGGG M01522:260:000000...
[7] 292 CCTACGGGATGCAGCAGTGGGGA...TTTAATCCTGATGAGCTAGAAA M01522:260:000000...
[8] 293 CCTACGGGGCGCAGCAGTAGGGA...TTAAAACTTTTGTTCTGGAATT M01522:260:000000...
[9] 292 CCTACGGGTTGCAGCAGTGGGGA...ATTAAAACTTTTCAGCTAGAGT M01522:260:000000...
[10] 293 CCTACGGGAGGCAGCAGTGGGGA...CCCGGGCTCAACCTGGGAACGG M01522:260:000000...And reverse reads should contain GACTACNVGGGTATCTAAT
Biostrings::readDNAStringSet(
fnRs[1],
format = "fastq",
nrec = 10
)DNAStringSet object of length 10:
width seq names
[1] 301 GACTACCAGGGTATCTAATCCTG...GGCTGCTGGCACGAAGTTCGCC M01522:260:000000...
[2] 301 GACTACCGGGGTATCTAATCCTG...GGCTGCTGGCACGGAGTTAGCC M01522:260:000000...
[3] 300 AATCCGGTTCGTGCCCCTAGGCT...TCTTTCCCAGCCCTTATTCCAA M01522:260:000000...
[4] 301 GACTACCGGGGTATCTAATCCTG...GGCTGCTGGCACGGAGTTAGCC M01522:260:000000...
[5] 301 GACTACCGGGGTATCTAATCCCT...GGCTGCTGGCCCGGAATTAGCC M01522:260:000000...
[6] 301 GGTATCTAATCCGGTTCGTGCCC...CACCGTCCTTACCCCCCCCTTT M01522:260:000000...
[7] 301 GGTATCTAATCTTGTTTGCTCCC...CCCGACGTTAGCCGGGGCTTCT M01522:260:000000...
[8] 301 GACTACGAGGGTATCTAATCCCG...GGCTGCTGGCACGGAATTAGCC M01522:260:000000...
[9] 301 GGTATCTAATCCTCTTCGCTACC...CACGAAGTTAGCCGGACCTTCT M01522:260:000000...
[10] 301 GACTACGGGGGTATCTAATCCTG...GGCTGCCGGCACGGGGTTAGCC M01522:260:000000...Use the function dada2::removePrimers() twice to remove the primers from forward fnFs and reverse fnRs reads and save the results in nopFs and nopRs respectively.
dada2::removePrimers(fn = fnFs,
fout = nopFw,
primer.fwd = primer_fwd,
max.mismatch = 1,
verbose = TRUE)
dada2::removePrimers(fn = fnRs,
fout = nopRv,
primer.fwd = primer_rev,
max.mismatch = 1,
verbose = TRUE)If you have to deal with a lot of samples and/or mixed orientated reads, we would recommend using dedicated tools such as cutadapt. We will see later during the course how to use cutadapt from the nextflow workflow nf-core/ampliseq.
Same as before, create a folder
path_to_filtered_reads <- here::here("outputs", "dada2", "filtered")
if (!dir.exists(path_to_filtered_reads)) dir.create(path_to_filtered_reads, recursive = TRUE)and list paths:
filtFs <- file.path(path_to_filtered_reads, basename(fnFs))
filtRs <- file.path(path_to_filtered_reads, basename(fnRs))To make the link between files and sample names, simply name vector of file names using the sample names
names(filtFs) <- sample_names
names(filtRs) <- sample_namesdada2::filterAndTrim()Let’s have a look at what the function is doing. To do so, type ?dada2::filterAndTrim() in the console.
Let’s run the function.
(out <- dada2::filterAndTrim(
fwd = nopFw,
filt = filtFs,
rev = nopRv,
filt.rev = filtRs,
truncLen = c(250,220),
minLen = 150,
matchIDs = TRUE,
maxN = 0,
maxEE = c(3, 3),
truncQ = 2
)) reads.in reads.out
S11B_R1.fastq.gz 1985 1318
S1B_R1.fastq.gz 1973 1367
S2B_R1.fastq.gz 1985 1385
S2S_R1.fastq.gz 1981 1321
S3B_R1.fastq.gz 1986 1352
S3S_R1.fastq.gz 1987 1452
S4B_R1.fastq.gz 1980 1393
S4S_R1.fastq.gz 1983 1444
S5B_R1.fastq.gz 1984 1358
S5S_R1.fastq.gz 1988 1394
S6B_R1.fastq.gz 1979 1327
S6S_R1.fastq.gz 1979 1347
S7B_R1.fastq.gz 1985 1328
S7S_R1.fastq.gz 1985 1371
S8B_R1.fastq.gz 1992 1306
S8S_R1.fastq.gz 1980 1352
S9B_R1.fastq.gz 1979 1283
S9S_R1.fastq.gz 1979 1344What happened?
Details about the function arguments: * nopFw : input, where the forward reads without primers are (path) * filtFs : output, where forward filtered reads are written (path) * nopRv and filRs : same as above, but with reverse reads * truncLen : truncate reads after truncLen bases. Reads shorter than that are discarded (truncLen=c(200,150) means forward and reverse reads are cut at 200 bp and 150 bp respectively) * trimLeft : number of nucleotides to remove from the start * trimright : number of nucleotides to remove from the end * maxN : maximum number of ambiguous bases accepted * maxEE : read expected errors (EE) threshold. The EE of a read is the sum of the error probability of each base composing it. Increase that value to accept more low quality reads. The first value refers to the forward reads and the second to the reverse reads. * truncQ=2: truncate reads at the first instance of a quality score less than or equal to truncQ.
To be able to denoise your data, you need an error model. The error model will tell you at which rate a nucleotide is replace by another for a given quality score. For example, for a quality score Q of 30, what is the probability of an A being wrongly read as a T.
This error model can be learnt directly from the data with the function dada2::learnErrors(). You can come back to the course for more details about the maths behind.
errF <- dada2::learnErrors(filtFs,
randomize = TRUE,
multithread = TRUE)6110500 total bases in 24442 reads from 18 samples will be used for learning the error rates.errR <- dada2::learnErrors(filtRs,
randomize = TRUE,
multithread = TRUE)5377240 total bases in 24442 reads from 18 samples will be used for learning the error rates.You can visualise the resulting error model using the function dada2::plotErrors()
dada2::plotErrors(errF, nominalQ = TRUE)Warning: Transformation introduced infinite values in continuous y-axis
Transformation introduced infinite values in continuous y-axis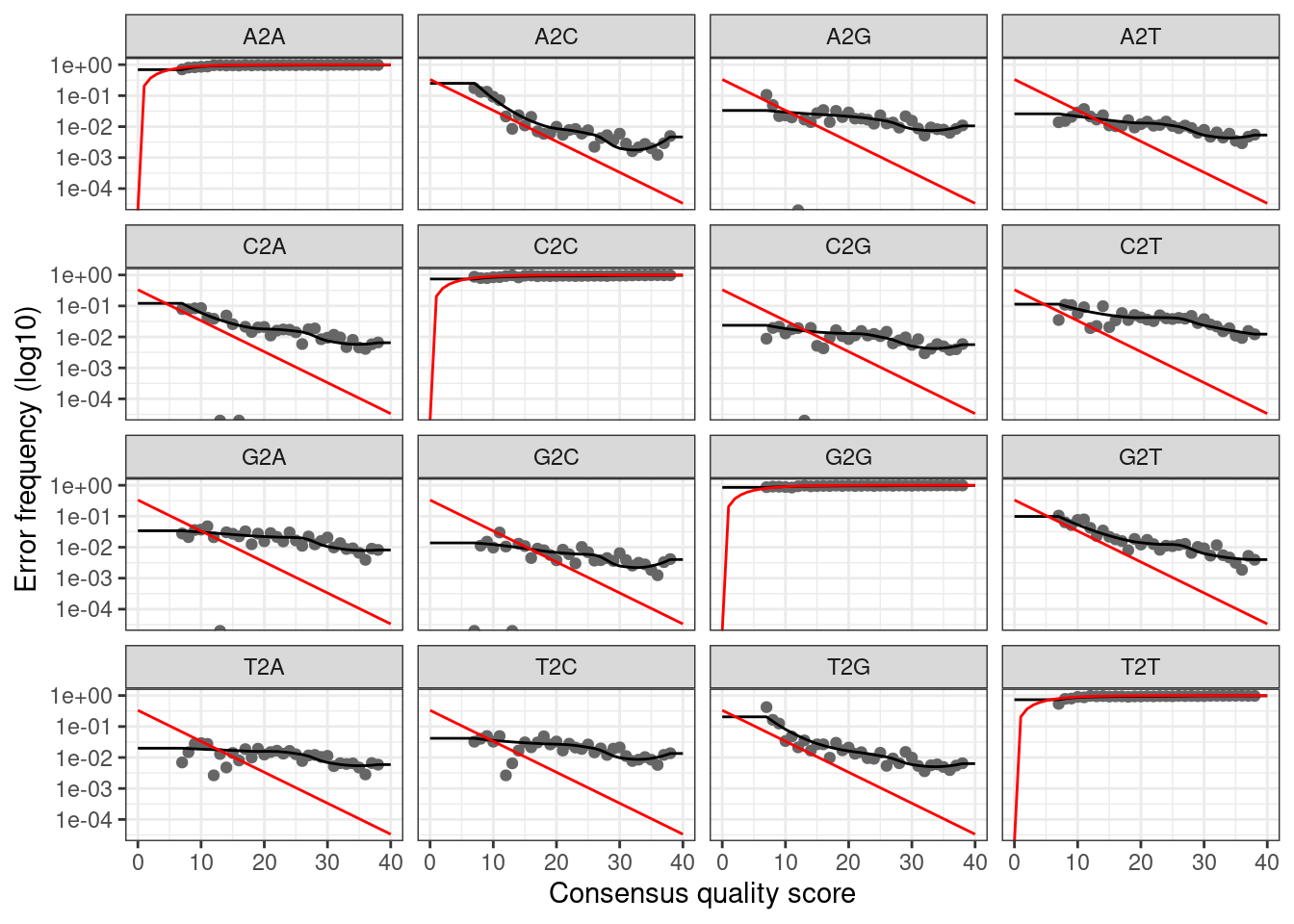
Before denoising, we need to dereplicate the sequences. It means, for each unique sequence, count the number of reads.
The dereplication is achieved using the function dada2::derepFastq()
derepFs <- dada2::derepFastq(filtFs, verbose = TRUE)
derepRs <- dada2::derepFastq(filtRs, verbose = TRUE)Now we are ready to run the denoising algorithm with dada2::dada(). As input, we need the error model and the dereplicated sequences.
dadaFs <- dada2::dada(derepFs, err = errF, multithread = TRUE)Sample 1 - 1318 reads in 793 unique sequences.
Sample 2 - 1367 reads in 843 unique sequences.
Sample 3 - 1385 reads in 814 unique sequences.
Sample 4 - 1321 reads in 799 unique sequences.
Sample 5 - 1352 reads in 803 unique sequences.
Sample 6 - 1452 reads in 801 unique sequences.
Sample 7 - 1393 reads in 756 unique sequences.
Sample 8 - 1444 reads in 668 unique sequences.
Sample 9 - 1358 reads in 807 unique sequences.
Sample 10 - 1394 reads in 701 unique sequences.
Sample 11 - 1327 reads in 698 unique sequences.
Sample 12 - 1347 reads in 697 unique sequences.
Sample 13 - 1328 reads in 730 unique sequences.
Sample 14 - 1371 reads in 757 unique sequences.
Sample 15 - 1306 reads in 735 unique sequences.
Sample 16 - 1352 reads in 734 unique sequences.
Sample 17 - 1283 reads in 707 unique sequences.
Sample 18 - 1344 reads in 710 unique sequences.dadaRs <- dada2::dada(derepRs, err = errR, multithread = TRUE)Sample 1 - 1318 reads in 815 unique sequences.
Sample 2 - 1367 reads in 834 unique sequences.
Sample 3 - 1385 reads in 872 unique sequences.
Sample 4 - 1321 reads in 799 unique sequences.
Sample 5 - 1352 reads in 832 unique sequences.
Sample 6 - 1452 reads in 851 unique sequences.
Sample 7 - 1393 reads in 838 unique sequences.
Sample 8 - 1444 reads in 746 unique sequences.
Sample 9 - 1358 reads in 852 unique sequences.
Sample 10 - 1394 reads in 777 unique sequences.
Sample 11 - 1327 reads in 734 unique sequences.
Sample 12 - 1347 reads in 710 unique sequences.
Sample 13 - 1328 reads in 754 unique sequences.
Sample 14 - 1371 reads in 816 unique sequences.
Sample 15 - 1306 reads in 788 unique sequences.
Sample 16 - 1352 reads in 814 unique sequences.
Sample 17 - 1283 reads in 751 unique sequences.
Sample 18 - 1344 reads in 782 unique sequences.Once forward and reverse reads have been denoised, we can merge them with dada2::mergePairs().
mergers <- dada2::mergePairs(
dadaF = dadaFs,
derepF = derepFs,
dadaR = dadaRs,
derepR = derepRs,
maxMismatch = 0,
verbose = TRUE
)At this point we have ASVs and we know their number of reads in each sample. We have enough information to build an ASV table.
seqtab <- dada2::makeSequenceTable(mergers)Chimeras are artifact sequences formed by two or more biological sequences incorrectly joined together. We find and remove bimeras (two-parent chimeras) using the function dada2::removeBimeraDenovo()
seqtab_nochim <- dada2::removeBimeraDenovo(seqtab,
method = "consensus",
multithread = TRUE,
verbose = TRUE)The ASV table is ready. But without a clue about the ASVs taxonomic identity, we won’t go far in our ecological interpretations. We can have an idea of ASV taxonomic identity comparing their sequences to references databases such as SILVA.
The taxonomic assignment is done in two steps.
First, each ASV is assigned to a taxonomy up to the genus level using the RDP Naive Bayesian Classifier algorithm described in Wang et al. 2007 called by the function dada2::assignTaxonomy(). We decided to assign at the genus level only because we consider the amplified 16S fragment not having a good enough resolution to assign at species level, except for perfect matches (see below).
taxonomy <- dada2::assignTaxonomy(
seqs = seqtab_nochim,
refFasta = silva_train_set,
taxLevels = c(
"Kingdom",
"Phylum",
"Class",
"Order",
"Family",
"Genus",
"Species"
),
multithread = TRUE,
minBoot = 60
)If you consider that in case an ASV is 100% similar to a reference sequence, it belongs to the same species, then you can use dada2::addSpecies() to add the species level.
taxonomy <- dada2::addSpecies(
taxonomy,
silva_species_assignment,
allowMultiple = FALSE
)This function assign to the species level ASVs which are identical to a reference sequence.
All the preprocessing is done. Now we export our results.
The results can be exported as a R objects, one object for the ASV table and another one for the taxonomy.
export_folder <- here::here("outputs", "dada2", "asv_table")
if (!dir.exists(export_folder)) dir.create(export_folder, recursive = TRUE)
saveRDS(object = seqtab_nochim,
file = file.path(export_folder, "seqtab_nochim.rds"))
saveRDS(object = taxonomy,
file = file.path(export_folder, "taxonomy.rds"))We recommand to export your results as text files. They are then reusable by other programs/languages.
But before, we need to format the data a little bit.
First we create a new variable to collect the ASV sequences:
asv_seq <- colnames(seqtab_nochim)We create unique ids for each ASV. The sequence itself is an unique id, but we would like to have something shorter.
ndigits <- nchar(length(asv_seq))
asv_id <- sprintf(paste0("ASV_%0", ndigits, "d"), seq_along(asv_seq))and rename the different variables with the new ids
row.names(taxonomy) <- colnames(seqtab_nochim) <- names(asv_seq) <- asv_idBefore exporting the data frames (taxonomy and seqtab_nochim) as a text file, we convert their row names (ASV ids) into a new column named asv. This is achieved using the custom function df_export()
taxonomy_export <- df_export(taxonomy, new_rn = "asv")
seqtab_nochim_export <- t(seqtab_nochim)
seqtab_nochim_export <- df_export(seqtab_nochim_export, new_rn = "asv")Finally, we can export the taxonomy
write.table(taxonomy_export,
file = file.path(export_folder, "taxonomy.tsv"),
quote = FALSE,
sep = "\t",
row.names = FALSE)the ASV table
write.table(seqtab_nochim_export,
file = file.path(export_folder, "asv_table.tsv"),
quote = FALSE,
sep = "\t",
row.names = FALSE)and the sequences as a fasta file
cat(paste0(">", names(asv_seq), "\n", asv_seq),
sep = "\n",
file = file.path(export_folder, "asv.fasta"))Statistics about each preprocessing step can also be exported.
First this table need to be assembled:
getN <- function(x) sum(dada2::getUniques(x))
log_table <- data.frame(
input = out[, 1],
filtered = out[, 2],
denoisedF = sapply(dadaFs, getN),
denoisedR = sapply(dadaRs, getN),
merged = sapply(mergers, getN),
nonchim = rowSums(seqtab_nochim),
perc_retained = rowSums(seqtab_nochim) / out[, 1] * 100
)
rownames(log_table) <- sample_namesThen it can be exported:
df_export(log_table, new_rn = "sample") |>
write.table(file = file.path(export_folder, "log_table.tsv"),
quote = FALSE,
sep = "\t",
row.names = FALSE)The sessionInfo() function in R provides a quick summary of your R environment, including the version of R, loaded packages, and their versions—useful for debugging, reproducibility, and sharing your setup with others.
sessionInfo()R version 4.3.1 (2023-06-16)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 22.04.3 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ANF_metaB_1.0.0
loaded via a namespace (and not attached):
[1] bitops_1.0-7 deldir_1.0-9
[3] remotes_2.4.2.1 rlang_1.1.1
[5] magrittr_2.0.3 matrixStats_1.0.0
[7] compiler_4.3.1 png_0.1-8
[9] callr_3.7.3 vctrs_0.6.3
[11] reshape2_1.4.4 stringr_1.5.0
[13] profvis_0.3.8 pkgconfig_2.0.3
[15] crayon_1.5.2 fastmap_1.1.1
[17] XVector_0.40.0 ellipsis_0.3.2
[19] labeling_0.4.2 utf8_1.2.3
[21] Rsamtools_2.16.0 promises_1.2.1
[23] rmarkdown_2.24 sessioninfo_1.2.2
[25] ps_1.7.5 purrr_1.0.2
[27] xfun_0.40 zlibbioc_1.46.0
[29] cachem_1.0.8 GenomeInfoDb_1.36.2
[31] jsonlite_1.8.7 later_1.3.1
[33] DelayedArray_0.26.7 BiocParallel_1.34.2
[35] jpeg_0.1-10 parallel_4.3.1
[37] prettyunits_1.1.1 R6_2.5.1
[39] stringi_1.7.12 RColorBrewer_1.1-3
[41] pkgload_1.3.2.1 GenomicRanges_1.52.0
[43] Rcpp_1.0.11 SummarizedExperiment_1.30.2
[45] knitr_1.43 usethis_2.2.2
[47] IRanges_2.34.1 httpuv_1.6.11
[49] Matrix_1.6-1 tidyselect_1.2.0
[51] rstudioapi_0.15.0 abind_1.4-5
[53] yaml_2.3.7 codetools_0.2-19
[55] miniUI_0.1.1.1 hwriter_1.3.2.1
[57] processx_3.8.2 pkgbuild_1.4.2
[59] plyr_1.8.8 lattice_0.21-8
[61] tibble_3.2.1 Biobase_2.60.0
[63] shiny_1.7.5 withr_2.5.0
[65] ShortRead_1.58.0 evaluate_0.21
[67] desc_1.4.2 RcppParallel_5.1.7
[69] urlchecker_1.0.1 Biostrings_2.68.1
[71] pillar_1.9.0 MatrixGenerics_1.12.3
[73] stats4_4.3.1 generics_0.1.3
[75] rprojroot_2.0.3 RCurl_1.98-1.12
[77] S4Vectors_0.38.1 ggplot2_3.4.3
[79] munsell_0.5.0 scales_1.2.1
[81] xtable_1.8-4 glue_1.6.2
[83] tools_4.3.1 interp_1.1-4
[85] GenomicAlignments_1.36.0 fs_1.6.3
[87] grid_4.3.1 latticeExtra_0.6-30
[89] devtools_2.4.5 colorspace_2.1-0
[91] GenomeInfoDbData_1.2.10 cli_3.6.1
[93] fansi_1.0.4 S4Arrays_1.0.5
[95] dada2_1.28.0 dplyr_1.1.2
[97] gtable_0.3.4 digest_0.6.33
[99] BiocGenerics_0.46.0 farver_2.1.1
[101] htmlwidgets_1.6.2 memoise_2.0.1
[103] htmltools_0.5.6 lifecycle_1.0.3
[105] here_1.0.1 mime_0.12